The genetic information needed to produce alpha chains is recorded in the 16th chromosome in humans.
Unlike for beta chains, there are four genes altogether coding for alpha chains, 2 on each copy of chromosome 16.
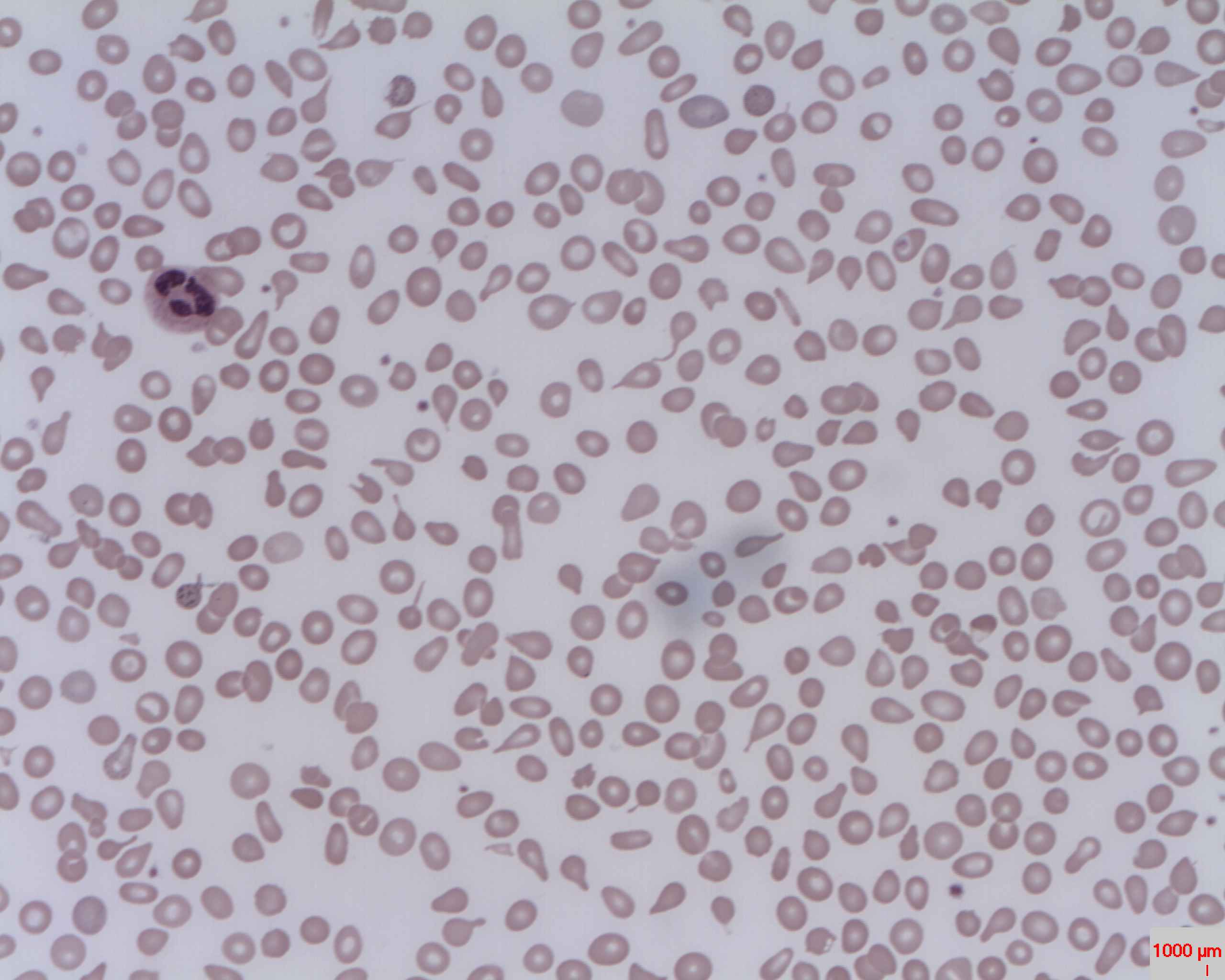
Alpha thalasaemia occur usually due to whole gene deletions.
Point mutations are rare, unlike in beta thalassaemia. According to how many genes get deleted, there are several kinds of thalassaemias.
- 1 or 2 genes deleted: alpha thalassaemia trait
- 3 genes deleted: HbH disease
- all 4 genes deleted: hydrops fetalis
Alpha thalassaemia trait is usually a symptomless condition requiring no treatment.
HbH disease, where only one alpha globin allele is functioning results in moderate to severe anaemia which might require blood transfusions occasionally.
Deletion of all 4 alleles is not compatible with life beyond the foetal stage.
Foetal death occurs. This condition is also called hydrops foetalis.
Unlike beta chains, alpha chains are a part of both HbA and HbF, hence it will affect the foetal stages of life as well.




No responses yet