The known trigger factors for crises like high altitudes and dehydration should be avoided, while maintaining good hygiene and nutrition, taking folic acid supplements if needed.
Vaccines for encapsulated bacteria are beneficial as the spleen might get affected.
Effective medical management options for the disease include hydroxyurea to increase fetal hemoglobin concentrations, opioids for pain management, and antibiotics for infections.
Sickle cell crises are managed with warmth and rehydration by oral fluids or saline until they resolve.
Severe cases of sickle cell anemia, however, might require blood transfusions or bone marrow transplants.
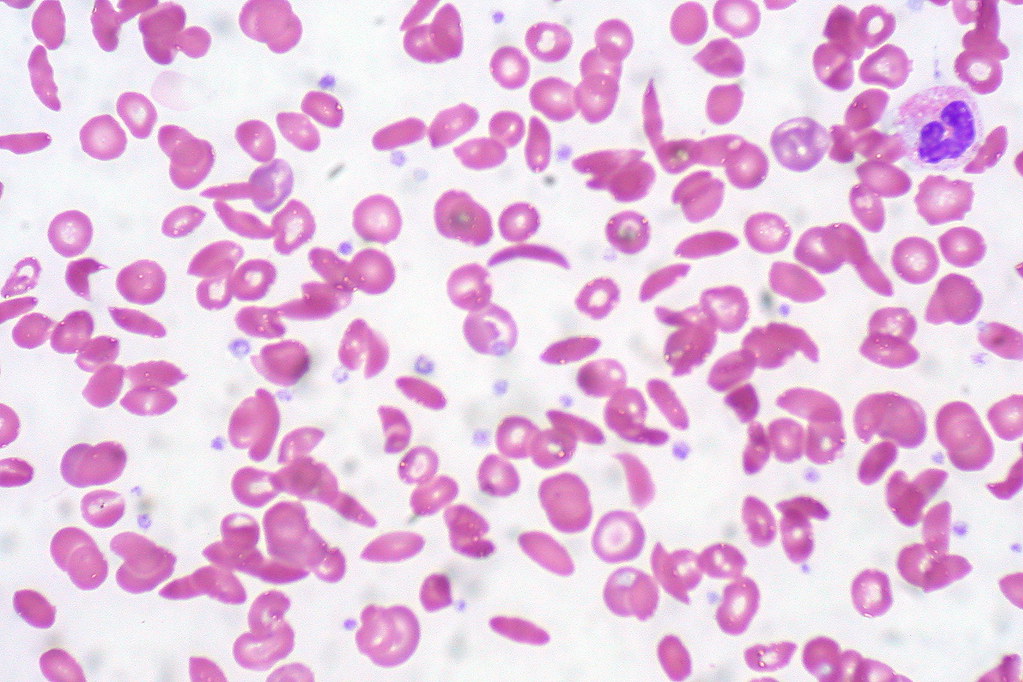



No responses yet